ウイルスの生態・進化と自然宿主
- ウイルスの自然宿主動物の同定と自然界における存続メカニズムの解明
- 鳥インフルエンザのグローバルサーベイランス
原子や分子レベルから地球レベルまで
ウイルスを理解する








ウイルスの世界を探究する最先端の研究室
私たちの研究室では、ウイルスの世界を原子や分子レベルの構造から地球レベルの生態まで幅広く探究しています。
具体的には、抗ウイルス薬の開発、細胞レベル・個体レベルでの感染メカニズムの解明、そして、ウイルスの自然宿主や伝播経路の解明やフィールドの疫学調査まで、いろいろな切り口でウイルスと感染症の研究に取り組んでいます。
このような多角的なアプローチにより、ウイルス性人獣共通感染症の制圧を目指し、ヒトと動物の健康を守るための研究を続けます。

近年、新興感染症が世界各地で発生しているが、それらのほとんどはウイルス性の人獣共通感染症である。人獣共通感染症を引き起こすウイルスの多くは、何らかの野生動物に感染し自然界に存続しており(そのような動物をそのウイルスの“自然宿主”と呼ぶ)、ヒトに伝播すると、時に致死率の高い感染症を引き起こす。したがって、自然宿主を突き止め、ウイルスの分布域や伝播経路を明らかにすることは、人への感染を防止するために極めて重要である。当研究室では、エボラウイルスやマールブルグウイルス等の出血熱ウイルスを対象に、アフリカにおけるウイルスの疫学調査を進めている。
A型インフルエンザウイルスは2種類の糖蛋白質ヘマグルチニン(HA)およびノイラミニダーゼ(NA)の抗原性によって亜型に分類され、これまでに16種類のHA(H1-H16)および9種類のNA (N1-N9)亜型のウイルスが本ウイルスの自然宿主である野生水禽から分離されている。現在までにパンデミックウイルスとしてヒトに流行したのはH1N1、H2N2およびH3N2であるが、近年これら以外の亜型の鳥インフルエンザウイルスのヒトへの感染が頻繁に報告されており、新たなパンデミックの可能性が危惧されている。特に高病原性鳥インフルエンザウイルスの拡散を監視するために、当研究室では日本、モンゴルおよびザンビアで、野生水禽を対象にウイルス保有調査を行ってきた。
ウイルス粒子の表面に存在するスパイク糖蛋白質は、宿主の標的細胞に存在する受容体分子(蛋白質や糖鎖等)と相互作用することで、ウイルスは細胞に吸着・侵入する。一般に、このようなウイルス蛋白質と受容体分子との特異的な相互作用は、ウイルスの組織特異性や宿主域を決定する重要な因子の一つである。また、ウイルスの表面蛋白質と中和抗体との相互作用も、ウイルスの抗原変異の理解およびワクチンや抗体医薬の開発において重要な知見となる。当研究室では、主に出血熱ウイルスの表面糖蛋白質の機能解析を通して、病原性発現および宿主域決定の分子基盤に関する研究を行っている。
抗体依存性感染増強(ADE)は、様々なウイルスで知られている。当研究室では、エボラウイルス、マールブルグウイルス、SARS-CoV-2に対する抗体の中にもADEを起こすものが存在すること事を示してきた。報告されているほとんどのADEは Fcレセプターを介したものであるが、詳細なメカニズムには不明な点が多い。これまでに、ADEにはFcレセプターは介在せず補体成分C1qが関与する経路の存在を明らかにしてきた。しかし、ADE抗体の存在意義には不明な点が多く、今後の解明が待たれる。
ウイルス蛋白質と宿主分子との相互作用は、ウイルス増殖過程やウイルス感染に対する生体防御応答において非常に重要である。当研究室では、分子シミュレーション等の計算科学的手法を活用し、ウイルス蛋白質と宿主分子の相互作用機序を原子・分子レベルで理解することを目指し研究を行っている。このような手法で、蛋白質の立体構造上でどのアミノ酸残基が分子間相互作用に重要であるかを詳細かつ定量的に調べることができるので、ウイルス蛋白質の特異性や機能を決めるアミノ酸残基の同定につながることが期待できる。

分子レベルで蛋白質の機能を理解するためには立体構造情報が必須である。しかしながら、世の中に存在するウイルスの数を考えると、その蛋白質の立体構造を全て実験的に決定するのは不可能である。そこで、当研究室ではコンピュータ上で立体構造を予測する分子モデリングの技術を駆使し、ウイルス蛋白質の構造解析を行っている。これまでに、インフルエンザウイルス、エボラウイルス、マールブルグウイルス、フラビウイルス、ブニヤウイルス、アレナウイルス等、数多くのウイルス蛋白質の立体構造を予測し、生物学的な実験を担当する研究者と共同で、ウイルスの抗原性や病原性、宿主特異性等の蛋白質機能に関わる因子の解明を行っている。
近年の構造生物学の進歩により、様々なウイルス蛋白質の立体構造データが公共データベースに多数登録されている。また、構造バイオインフォマティクスの発展により、計算機上で構造未知の蛋白質の立体構造を予測する手法や立体構造から蛋白質の機能を推定する手法等、構造解析技術の整備が急速に進んでいる。当研究室では、ウイルス蛋白質の構造や配列情報を詳細かつ大規模に比較解析し、ウイルス蛋白質の未知の機能部位を推定する手法の開発を行っている。
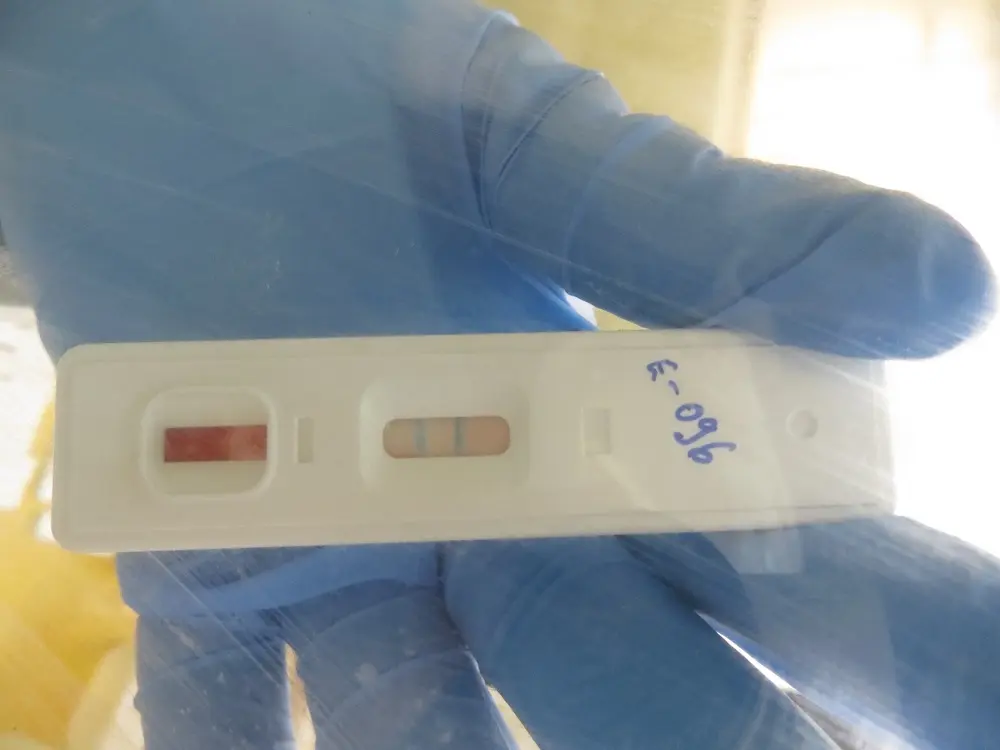
エボラウイルスやマールブルグウイルス等の出血熱ウイルスおよびインフルエンザウイルスの構造蛋白質に対すモノクローナル抗体を多数作出してきた。それらを用いて、基礎研究に活用するとともに、予防・治療法および診断法の開発を行っている。例えば、抗体によるウイルス中和のメカニズム解明やウイルス種間あるいは株間で広く交差反応性を示す抗体の探索を行っている。
エボラおよびマールブルグウイルスなどのフィロウイルスに対して認可された治療薬は、長年にわたり存在しなかった。しかし近年、エボラ出血熱に対する抗体医薬Inmazeb(2020年10月認可)およびEbanga(2020年12月認可)がアメリカの食品医薬局(FDA)により承認され、エボラ出血熱の治療に光が見えてきた。しかし、これらの抗体医薬は、出血熱の病原体として知られている5種のフィロウイルスのうち、たった1種のウイルスに対してしか効果がない。将来のアウトブレイクに備えるため、様々なフィロウイルスに幅広く効果を示す治療薬の開発が必要である。当研究室では、複数のフィロウイルスに対して幅広く効果を示す抗体の作出や化合物・天然物のスクリーニングを通して、エボラおよびマールブルグ出血熱に対する汎用性の高い治療薬開発を目指し、研究を行っている。
エボラやマールブルグウイルス出血熱の診断には主にリルタイムPCR法などが使用されており、特別な装置と比較的長い測定時間を必要とする。したがって、都市から離れた地域で感染者が発生した場合には迅速に診断ができず、感染拡大の要因となっていた。そこで、当研究室ではデンカ(株)と共同で、イムノクロマト法(エボラおよびマールブルグウイルスの核蛋白質に対する抗体を使用)を用いた診断キットを開発し、エボラ出血熱がコンゴ民主共和国で発生した際に提供している。この診断キットは、特別な器具や装置を必要とせず、室温で長期間安定、簡便且つ迅速な検査が可能で、電源などが十分でない地域においても活用可能である。
コンピュータを利用した医薬品設計は、大きく分けるとLigand-based drug-design(LBDD)とStructure-based drug-design(SBDD)の2つに分類される。LBDDは主に既知のヒット化合物の情報を用いて行うもので、SBDDは主にタンパク質の立体構造情報を用いて行うものである。SARS-CoV2やインフルエンザウイルスなど、種々のウイルスを対象に、バーチャルスクリーニングを活用した薬剤探索を行い、抗ウイルス作用を示す化合物を見出している。また、立体構造情報を起点とした抗ウイルス性ペプチドの設計などにも取り組んでいる。