
Introduction to the Research Activities at Our Laboratory
At our laboratory, we carry out fundamental research on viral diseases.
As many of the members at our laboratory have studied pathology, we are good at immunocytochemical and immunohistochemical methods. In addition, we usually apply molecular biological and virological experiment techniques to various experiments.
Our research is based on three pillars, namely, 1) Analysis of pathological mechanisms of virus-infected hosts, 2) Analysis on the intracellular kinetics of viruses, and 3) Development of treatment methods for virus diseases.
We are currently involved in the following research projects.
Pathological Analysis of West Nile Virus Disease
Flaviviruses include a number of zoonoses, and can be transmitted to and cause illness in both humans and animals through arthropods. We have been studying flavivirus diseases focusing on West Nile Virus (WNV), which can cause severe encephalomyelitis, as a model. WNV diseases are prevalent mainly in the United States and Europe, but there is concern that such diseases may enter and spread throughout Japan as well.
WNV travels from the periphery to brain tissue, and replicates in neuronal cells in the central nerve system (CNS). neuronal death with viral replication plays a major role in the pathogenesis of the virus. To identify the mechanism of cerebral infiltration of WNV, we measured the permeability of WNV in vascular endothelial cells. Then, the permeability of a virulent WNV strain (NY99 strain), which causes encephalitis in mice by peripheral inoculation, and that of an attenuated strain (Eg101 strain) were compared. High permeability of the NY99 strain in the vascular endothelial cells was observed when there was a difference between the two amino acids of E protein that form an envelope (Hasebe et al, BMC Microbiol, 2010).
Next, changes of neuronal cells in WNV-infected mice were analyzed histopathologically and biochemically. The accumulation of insoluble ubiquitinated proteins in the WNV-infected neuronal cells was then observed. The accumulation of insoluble ubiquitinated proteins is known to occur in neurodegenerative diseases causing neuronal death. The obtained results suggest that neuronal cell death during WNV infection is caused by the accumulation of insoluble ubiquitinated proteins (Figure 1) (Kobayashi et al, Neuropathology, 2012).
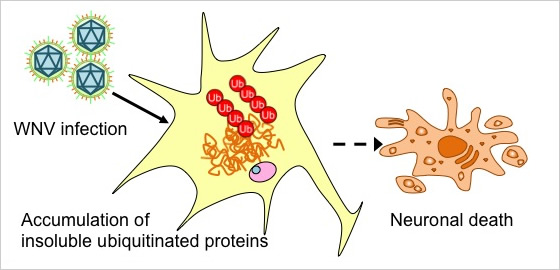
Figure 1.
We also sought to understand the role of autophagy in WNV replication, focusing on autophagy as a host response to various viral infections. Following WNV infection, expression of LC3-II, a marker for autophagy, increased in human neuroblastoma. WNV replication in cells lacking Atg5, and essential factor for autophagy, was increased. From the above results, autophagy is shown to be induced by WNV infection and inhibit WNV replication (Kobayashi et al, Virus Res., 2014).
Joint research by Professor Niikura from the Research Institute for Electronic Science, Hokkaido University, Professor Suzuki from the National Institute of Infectious Diseases and others could show that the production of WNV antibodies increased using gold nanoparticles. Host responses such as cytokine release varied depending on the shape and size of the gold nanoparticles (Niikura K et al, ACS Nano, 2013).
Furthermore, the mechanism of transcellular transport of virus particles in the process of intracellular WNV infiltration was examined in detail. During infiltration, movement of virus particles was slow in the early stage, and faster in the late stage. Using fluorescently-labeled virus-like particles, this fast movement in the late stage was shown to be microtubule-dependent (Makino et al, J Virol Methods, 2014).
Currently, we are focusing our research on the identification of the intracellular kinetics of WNV and its extracellular release mechanism.
Research toward the Development of Treatment Method to Neutralize Virus Diseases
Currently, we are carrying out joint research with SHIONOGI & Co., Ltd. for the development of treatment methods for viral diseases, especially dengue fever.
Dengue fever is an infectious disease caused by the dengue virus (DENV), of the flaviviridae family. It is mainly Aedes spp, and the infection cycle is established among humans. DENV is divided into 4 serotypes (DENV-1, DENV-2, DENV-3 and DENV-4). The cause of the dengue fever outbreak in Tokyo’s Yoyogi park in 2014 is reported to be DENV-1. Dengue fever occurs in over a hundred of tropical and subtropical countries in Asia, Americas and Africa. It has been reported that more than 1 billion people around the world are in danger of infection. Humans infected with DENV exhibiting viremia, serve as a supply source of the virus to other humans. Dengue fever occurs in 20-50% of those infected with DENV, and cures in about a week. Infection by other serotypes of DENV after initial infection increases the risk of severe dengue with plasma leakage and bleeding tendency.
Currently, prevention and treatment methods for infection have not been established. We will continue to work towards the development of therapeutic drugs through this joint research. Joint research for other emerging and re-emerging infectious diseases is also being planned.
Analysis on the Cell Infection Mechanism in Rabies Virus
Rabies is a zoonosis which causes over 55,000 human deaths around the world each year. Rabies virus (RABV) is the causative virus of rabies. It can be transmitted to a wide range of mammals. It invades the human body through a bite from a rabies-infected animal, and causes fatal neurological symptoms when it reaches the central nerve system.
In order to identify the mechanism of cell multiplication of the rabies virus, we are investigating possible intracellular factors which are involved in the multiplication of rabies virus and those which are involved in the inhibition of virus multiplication. We also are examining the interaction between virus-encoded proteins and intracellular factors in detail.
In our joint research with Professor Sugiyama and Professor Ito from the Faculty of Applied Biological Sciences, Gifu University, we have produced recombinant RABV using reverse genetics. Currently we have confirmed the characteristics of the produced recombinant RABV, and are planning to develop a method for inhibiting rabies in the future.
Identification of Intracellular Replication Mechanism of the JC Virus
JC Virus is a non-enveloped double-stranded DNA virus with a size of approximately 40 nm in diameter, belonging to the polyomavirus family. Most humans are infected by JCV in childhood. Seventy to eighty % of normal adults have antibodies against JCV. However, in cases of immunodeficiency such as AIDS, JCV can multiply mainly in oligodendrocytes in the brain and cause fatal progressive multifocal leukoencephalopathy (PML). So far, no effective treatment method for PML has been established.
To identify the intracellular multiplication mechanism in JCV, we have analysed the JCV-encoded protein function and genome expression mechanism, and examined host factors which regulate JCV replication, in a joint study with Dr. Suzuki from the National Institute of Infectious Diseases (Tokyo, Japan). Based on Obtained results, we hope to discover the treatment method for the inhibition of JCV replication and a cure for PML. The intracellular replication mechanism of JCV which we have identified up to now is shown below. (Figure 2). The latest findings are introduced below.
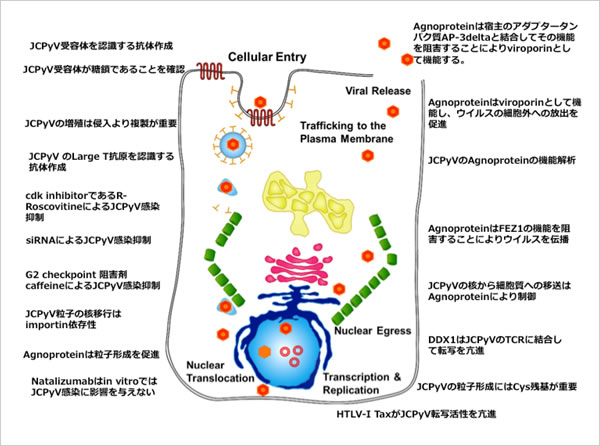
Figure 2. Intracellular multiplication mechanism of JCV we have identified up to now (Virus 64:1: pp. 25-34, 2014 in Japanese)
Cell lysis is necessary for cell egress of a non-enveloped virus. Prior to cell lysis, permeability of cell membrane to small molecules such as ions is known to increase. This increase in permeability of the cell membrane is considered to be the cause of cell destruction. Recently, viral proteins were found to directly affect the induction of increase of cell membrane permeability. Regarding this new concept, classification of the group of the viral proteins which possess this function as viroporins, has been proposed.
In JCV, a late protein, agnoprotein, was found to function as a viroporin (Suzuki T. et al. PLoS Pathog. 2010). Agnoprotein forms homooligomers on the cell membrane, increasing permeability of the cell membrane and inducing cell lysis, and promoting cell egress of virus (Figure 3).
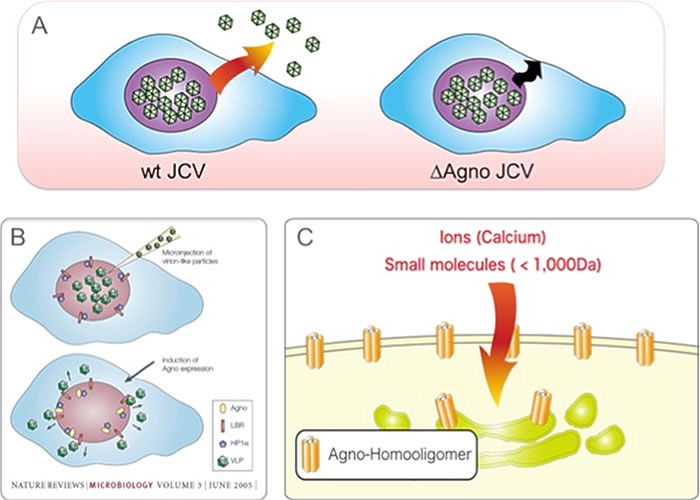
Figure 3. A) JCV virions are generated in the nuclei and egressed from the cell. Agnoprotein deletion mutant virus (ΔAgno JCV) fails to release particles from the cells. B) Agnoprotein interacts with HP1α and promotes transportation of virus particles from the nuclei to the cytoplasm. C) Agnoprotein forms homooligomers on the cell membrane and functions as a viroporin. Viroporin induces cell lysis and promotes extracellular release of virus particles.
Also, agnoprotein binds to heterochromatin protein HP1α on the nuclear membrane. It has been found that this interaction changes the structure of the nuclear membrane, promoting the transportation of virus particles from the nuclei to the cytoplasm (Okada Y. et al. EMBO Rep. 2005). In order to discover the molecular mechanism where agnoprotein functions as viroporin, we used a yeast two-hybrid system to search for a host factor that binds to agnoprotein. As a result, AP3D, the δ subunit of adaptor protein complex 3 (AP3) involved in intracellular substance trafficking, was identified as an agnoprotein-binding factor. The interaction between the identified AP3D and agnoprotein was analyzed in detail using molecular biological and biochemical techniques. The results showed that agnoprotein directly binds to AP3D and inhibits the intracellular vesicular trafficking system regulated by AP3, inhibiting the trafficking of agnoprotein itself to the lysosomal degradation system. As a result, agnoprotein can be transported to the cell membrane without being degraded in lysosomes, functioning as a viroporin (Figure 4). Furthermore, based on the obtained results, the agnoprotein-binding site of AP3D was shown to be over-expressed, and extracellular release of virus particles was inhibited. Consequently JCV infection could be inhibited. These results show that interaction with the host factor is essential for the expression of viroporin function, and that interface of the interaction of viroporin-host factor can be targeted for therapeutic drugs for virus infections. The same concept can be applied not only for polyomaviruses but also for other viruses containing viroporin such as influenza, and the development of therapeutic drugs for viral infections targeting viroporin is expected (Suzuki T. et al. Proc Natl Acad Sci USA, 2013).
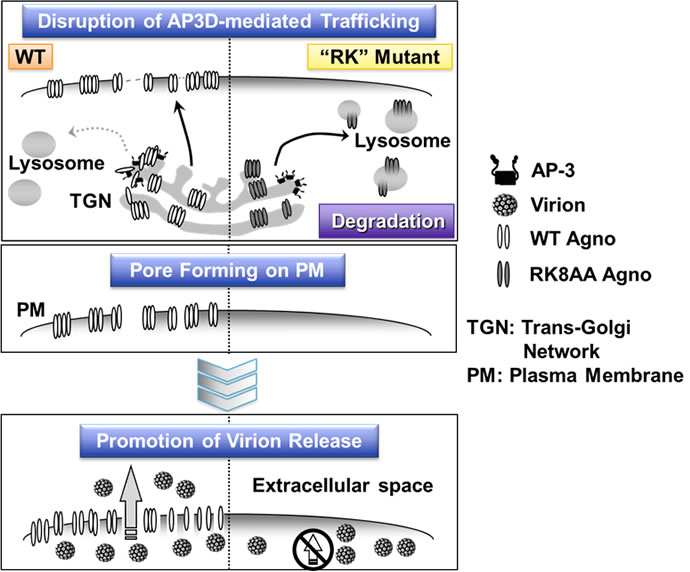
Figure 4. Agnoprotein in JCV directly binds to the host factor AP3D, blocking the trafficking of agnoprotein itself to lysosomes and inhibiting degradation. Agnoprotein is transported to the cell membrane and forms polymers, functioning as a viroporin and promoting extracellular release of JCV particles. However, agnoprotein which contains RK mutant and which is non-binding to AP3D, is degraded by lysosomes.